Results
To investigate the affect of mutations on cld function, our workflow involved three stages: Alignment, Structure, and Stability
Project Overview
1. Alignment
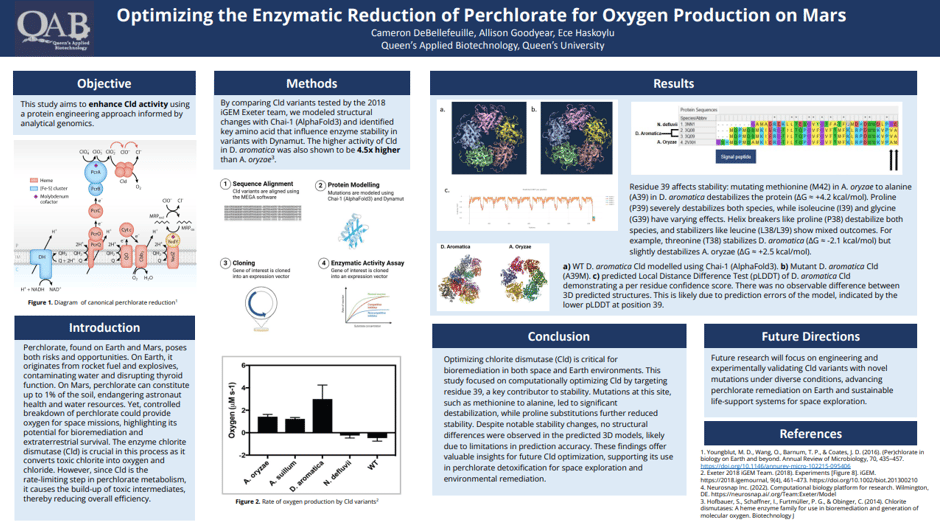
The 2018 Exeter iGEM Team found a substantial difference in oxygen production between different variants of chlorite dismutase (Cld). Specifically, the Cld from D. aromatica had a significantly higher turnover rate than that of A. oryzae, which is the variant used by the NASA Ames research team. Our goal was to investigate why D. aromatica had a much higher turnover rate than A. oryzae. To achieve this, we performed a sequence alignment of all the enzymes used by the 2018 Exeter team to identify amino acid differences. We then analyzed the impact of the identified amino acid substitutions, and finally, we modeled how de novo mutations affected the protein structure.
Figure 1. Sequence alignment using the Mega software. The cld variants are identified using their accession numbers, with 3NN1 representing the variant from N. defluvii, 3Q08 and 3Q09 from D. aromatica, and 2VXH from A. oryzae


From the alignment, we found that there were no differences in the active site, which consists of a heme cofactor, H170 in the proximal pocket, and R183 in the distal pocket. Instead, the only amino acid differences were identified at positions 38 and 39 in D. aromatica, where valine (V38) and alanine (A39) were present. Notably, V38 stood out as a potential factor influencing enzyme activity, as valine is known to act as a helix breaker. This disruption in secondary structure could lead to conformational changes that enhance catalytic efficiency. To investigate this, we analyzed the structural implications of these substitutions through molecular modeling, assessing how they might alter enzyme dynamics and function.
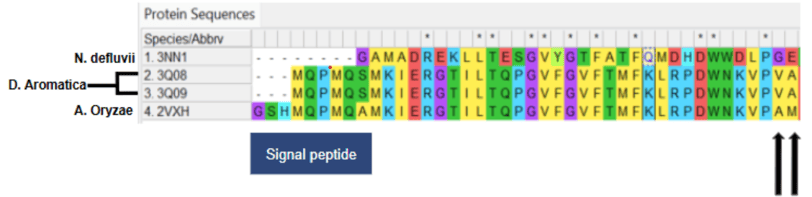
Figure 2. Annotated sequence alignment with the signal peptide, variant, and substitutions identified
2. Structure
3. Stability
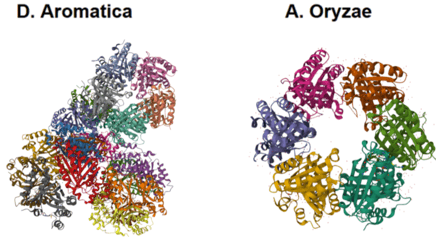
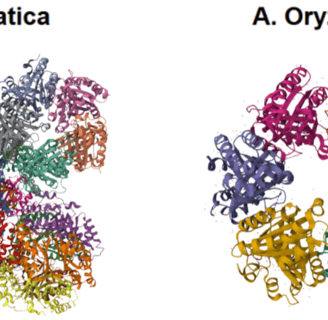
Figure 4. Structures from PDB of D. aromatica and A. oryzae. D. aromatica is a pentamer which forms an asymmetric unit of 20 pentamers, while A. oryzar is a hexamer.
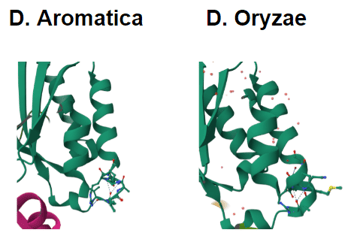
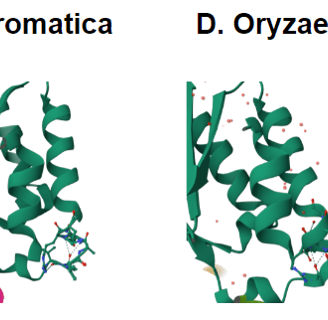
Figure 3. Structures from PDB of D. aromatica and A. oryzae. D. aromatica revealing the structural differences where the amino acid differences are.
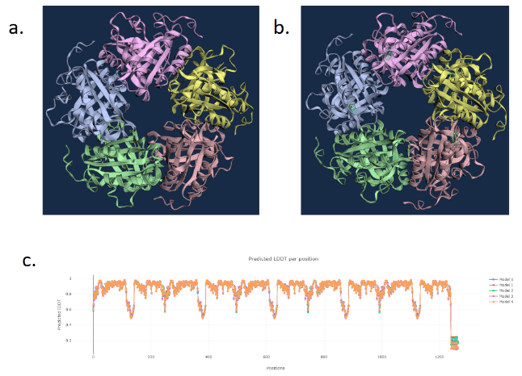
Chai-1 Modelling
Figure 5. a) WT D. aromatica Cld modelled using Chai-1 (AlphaFold3). b) Mutant D. aromatica Cld (A39M). c) predicted Local Distance Difference Test (pLDDT) of D. aromatica Cld demonstrating a per residue confidence score. There was no observable difference between 3D predicted structures. This is likely due to prediction errors of the model, indicated by the lower pLDDT at position 39.
Boltz-1 (AlphaFold3) Modelling
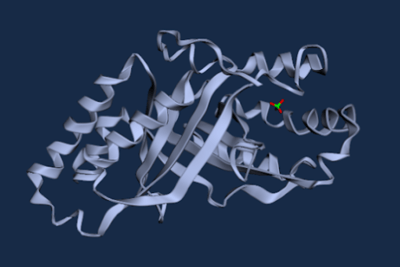
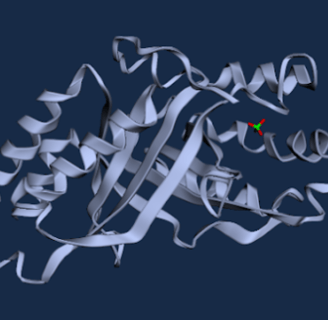
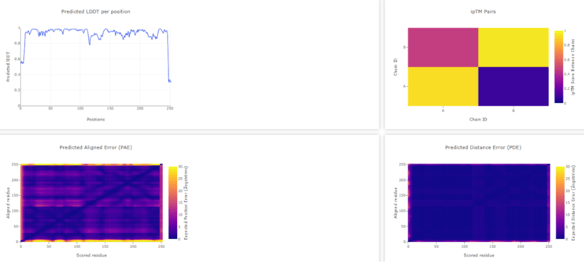
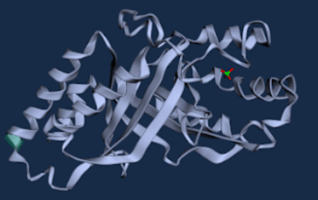
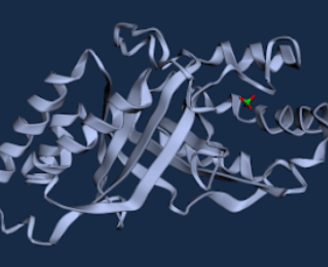
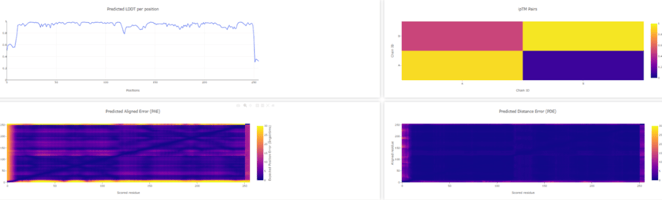
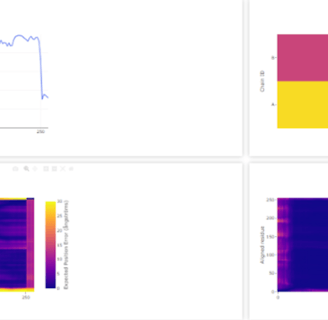
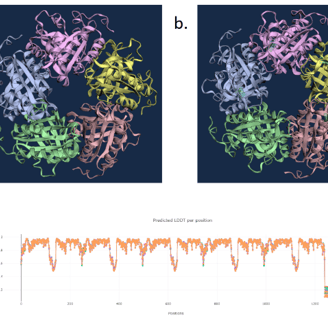
Figure 6. WT D. aromatica Cld modelled using Boltz-1 (left). Predicted Local Distance Difference Test (pLDDT) of D. aromatica Cld demonstrating a per residue confidence score (right).
Figure 7. WT A. oryzae Cld modelled using Boltz-1 (left). Predicted Local Distance Difference Test (pLDDT) of D. aromatica Cld demonstrating a per residue confidence score (right).
Our structural modeling using Chai-1 (AlphaFold3) and Boltz-1 showed no significant differences between WT D. aromatica Cld and its mutant variants, despite amino acid substitutions observed in crystal structures. This discrepancy suggests that current prediction models may lack the accuracy needed to capture subtle but functionally relevant structural variations. The inability to resolve these differences highlights the limitations of AI-based modeling in cases where minor sequence changes may have significant effects on enzyme dynamics and activity. Future studies using experimental validation, such as molecular dynamics simulations or crystallographic analysis, would be essential to confirm the structural impact of these amino acid variations.
To investigate structural differences, we analyzed D. aromatica and A. oryzae Cld using PDB data. Figure 3 highlights key amino acid differences, while quaternary structures also differ—D. aromatica forms pentamers assembling into larger units, whereas A. oryzae is a hexamer (Figure 4). We modeled WT and mutant D. aromatica Cld using Chai-1 (AlphaFold3) and Boltz-1. Figure 5 compares WT and A39M mutant structures predicted by Chai-1, showing no significant differences, likely due to model prediction limitations. Additional Boltz-1 predictions (Figures 6 and 7) further assess structural and functional impacts of sequence variations.
Finally, we leveraged Dynamut, a bioinformatic software that models the stability of proteins based on their amino acid sequence. This was particularly relevant to us because we were able to look at the alino acid alignments between D. aromatica and A. oryzae and model how the few amino acid differences they have influence their stability. Residues 38 and 39 were of special interest, and it was observed that mutating the alanine at residue 39 in D. aromatica to a methionine (like in A. oryzae), the protein was destabilized at a large scale (Figure 8.) relative to the effects of other mutations.


To futher the scope of the analysis, in addition to making the specific mutations that differed D. aromatica and A. oryzae, many different amino acids were mutated at the two residues of interest. We also wanted to explore how added heliz breakers could influence stability. Some amino acid mutations had the same effect on both species, such as proline and serine (helix breakers) both causing destabilized at residues 38 and 39. Other amino acids had different influences based on the residue or species. When glycine (heliz breaker) or asparagine (weaker helix breaker) were mutated in, they both stabilized the base species at residue 39, but caused destabilization at residue 38. Isoleucine (helix breaker) had the opposite effect, destabilizing at residue 39 and stabilizing at residue 38.
Other amino acids showed varying trends, and certain helix breakers resulted in stabilization, demonstrating that the addition of a helix breaker does not necessarily determine whether the protein will stabilize or destabilize, as so many other protein interactions contribute to stability.